

Построение 3D-моделей нециклических молекул в естественных переменных
Построение 3D-моделей нециклических молекул в естественных переменных
Е.Г. Атавин, В.О. Тихоненко, Омский государственный университет, кафедра органической химии
1. Введение
По мере накопления химической информации роль данных о пространственном геометрическом строении молекул возрастает. Устанавливать его можно как экспериментальными, так и теоретическими методами, а описывать принято либо в декартовой системе координат, либо в естественных (внутренних, молекулярных) переменных.
Первый способ предполагает знание 3N декартовых координат N атомов, позволяет легко строить графическое изображение молекулы, вычислять значения всех естественных переменных и используется в большинстве современных программ квантовой механики, молекулярной механики и колебательной спектроскопии. Однако произвол в выборе положения начала координат и ориентации координатных осей затрудняет сравнение результатов, полученных разными авторами. Кроме того, наличие у молекулы трех поступательных и трех вращательных степеней свободы приводит к появлению шести нулевых собственных значений у матрицы вторых производных энергии по координатам и к дополнительным осложнениям вычислительного характера [1]. Наконец, само задание декартовых координат атомов - нетривиальная задача, поскольку они не являются справочными данными.
Описание (и анализ) геометрического строения в естественных переменных (ниже - межъядерные расстояния R, валентные углы , , и углы внутреннего вращения , F) проще, поскольку задание их не представляет проблемы и менее зависит от произвола исследователя, благодаря имеющимся эмпирическим закономерностям [2]. При оптимизации геометрии молекулы можно упрощать задачу, фиксируя значения хорошо известных параметров. Легко организовать поиск глобального минимума энергии путем перебора допустимых значений всех или некоторых параметров. При работе же с декартовыми координатами реализация этих возможностей сопряжена со значительными трудностями.
Однако непосредственно по значениям естественных переменных невозможно в общем случае построить графическое изображение молекулы. Также затруднительно выполнять любые вычислительные операции с моделью молекулы, например, определять вандерваальсовые расстояния между атомами.
Таким образом, оба способа описания молекулярной геометрии обладают рядом практически важных достоинств и весьма существенных недостатков. Совмещение достоинств достигается вычислением декартовых координат атомов по заданным естественным переменным, что представляет собой в общем случае весьма громоздкую стереометрическую задачу.
Цель настояшей работы и состоит в рассмотрении алгоритмов вычисления декартовых координат атомов по заданным естественным переменным то есть построения 3D-моделей молекул.
2. Метод Эйринга [3]
Обычно систему координат связывают с положением первых трех атомов (рис. а), координаты которых, таким образом, определяются по формулам:
|
x1 = R12 cos, |
x2 = 0, |
x3 = R23 |
|
|
y1 = R12 sin, |
y2 = 0, |
y3 = 0 |
(1) |
|
z1 = 0, |
z2 = 0, |
z3 = 0 |
Легко также вычислить координаты четвертого атома:
|
x4=x3-R34cos |
|
|
y4=R34sincos |
(2) |
|
z4=R34sinsin |
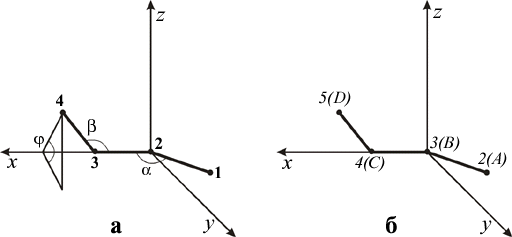
Рис. 1. Ориентация молекулы в системе координат
Далее, построенный фрагмент с помощью переноса и двух поворотов переводится в положение, показанное на рис. 1б.
|
|
|
|
|
(i - номера всех ранее построенных атомов), что дает возможность вычислить координаты следующего атома по формулам (2) и т.д. Общее число умножений и делений при построении модели N-атомной молекулы растет квадратично и составляет 6+4N·(N-4) операций.
3. Алгоритм построения моделей больших молекул
В предлагаемом алгоритме отсутствуют многократные переносы и вращения ранее построенных фрагментов, новые атомы встраиваются в растущую цепь непосредственно, без ее предварительной переориентации,что, помимо увеличения быстродействия, более благоприятно с точки зрения устойчивости вычислительной схемы к накоплению ошибок округления.
Координаты первых четырех атомов вычисляются по формулам (1, 2).
Выбираются три атома A, B, C с найденными координатами (Xi,Yi ,Zi ), где i = a, b, c .
Переносим систему координат в точку опорного атома B:
|
Вычисляем координаты атомов A, B, C и пристраиваемого атома D во вспомогательной системе координат по формулам (1, 2).
Полученные координаты связаны ортогональным преобразованием A
|
|
|
элементы которого для случая собственного вращения (Det(A) = 1) удается выразить следующим образом:
|
|
|
|
|
|
(случай хc = Rbc = 0 в молекулах не встречается; случай уa = Rab sin = 0 возникает в производных ацетилена и легко исключается выбором в качестве атомов A, B и C другого, нелинейного фрагмента).
Лишь три из девяти матричных элементов aij независимы. Справедливость связывающих их условий, накладываемых ортогональностью линейного преобразования А, может быть проверена непосредственно.
Координаты атома D (xd , yd , zd ) преобразуются в исходную систему координат:
|
и процесс повторяется с пункта 2 до полного построения модели. Общее число умножений и делений растет линейно и может быть уменьшено до 6+27·(N-4) операций.
Отношение числа операций в алгоритме Эйринга и в предлагаемой схеме близко к N/7, превышает единицу уже для семиатомных цепей, а для молекул большого размера оказывается весьма значительным.
Отметим, что матрица A является общей для различных атомов D, что, в частности, значительно ускоряет вычисление координат атомов водорода.
4. Метод Нордландера
Вместо линейного преобразования А переход от вспомогательной системы координат к системе, связанной с молекулой, можно осуществить с помощью трех последовательных вращений вокруг координатных осей. Однако соответствующие формулы [4] выглядят существенно более громоздко, требуют постоянного выбора оптимального решения из разных вариантов последовательностей вращений и требуют не менее 6+55·(N-4) операций умножения и деления и 3·(N-4) операций извлечения корня.
5. Метод Эдди
Весьма эффективным является алгоритм [5], использующий тот факт, что координаты атома D легко выражаются через направляющие косинусы связи CD. Последние связываются с направляющими косинусами двух предыдущих связей и структурными параметрами.Число умножений и делений составляет 8+36·(N-4) операций. Приходится хранить дополнительно 3·(N-2) значений косинусов.
6. Разветвленные цепи
Методика построения боковых цепей не отличается от построения главной цепи, необходимо лишь соответствующим образом задавать последовательности опорныx атомов A, B, C и молекулярных параметров. Известно, однако,

Рис. 2. Построение боковых цепей
что точность задания торсионных углов по справочным данным на порядок ниже, чем валентных, и значительно более точное описание взаимного расположения связей 3-4 и 3-5 узлового атома 3 (рис. 2) достигается заданием не двух торсионных углов 1234 и F1235, а одного торсионного 1234 и одного валентного 435 (помимо обязательных для обоих вариантов валентных углов 234 и 235) [6]. Вычислить требуемый торсионный угол F можно из соотношений:
|
|
|
|
Выбор знака + или - определяется желаемой (L или D) конфигурацией узлового атома.
В особом случае разветвления у второго атома (например в изобутане) для определения угла F удобно ввести в качестве опорного атома А вспомогательный атом с координатами
|
|
|
Тогда = 180, cosF = - С, sinF = S.
Список литературы
Hilderbrandt R.L. Application of Newton-Raphson optimization techniques in molecular mechanics calculations// Computers & Chemistry 1977 V. 1. P. 179-186.
Mastryukov V.S., Simonsen S. H. Empirical correlations in structural chemistry // Molecular Structure Research. 1996. V. 2. P. 163-189.
Дашевский В.Г. Конформационный анализ органических молекул М.: Химия, 1982 .
Nordlander J.E., Bond A.F., Bader M. Atcoor: a program for calculation and utilization of molecular atomic coordinates from bond parameters// Computers & Chemistry. 1985. V. 3. P. 209-235.
Eddy C. R., Computation of the Spatial Locations of Atoms of a Chain Molecule Undergoing Intramolecular Rotations// J. Chem. Phys. 1963. V. 38. P. 1032-1033.
Зенкин А.А. Алгоритмы построения 3D-моделей молекулярных систем // Тезисы IX Всесоюзной конференции ;Химическая информатика; Черноголовка. 1992. Ч. 1. С. 8.
Для подготовки данной работы были использованы материалы с сайта http://www.omsu.omskreg.ru/
Дата добавления: 05.12.2006
